
La secuencia genética de 17 genomas del coronavirus confirma que las cepas de la epidemia en el país provienen de Europa y Estados Unidos; los grupos provenientes de Asia sólo permanecieron dos meses
Texto: Arturo Contreras Camero y Daniela Pastrana
Foto: María Ruiz
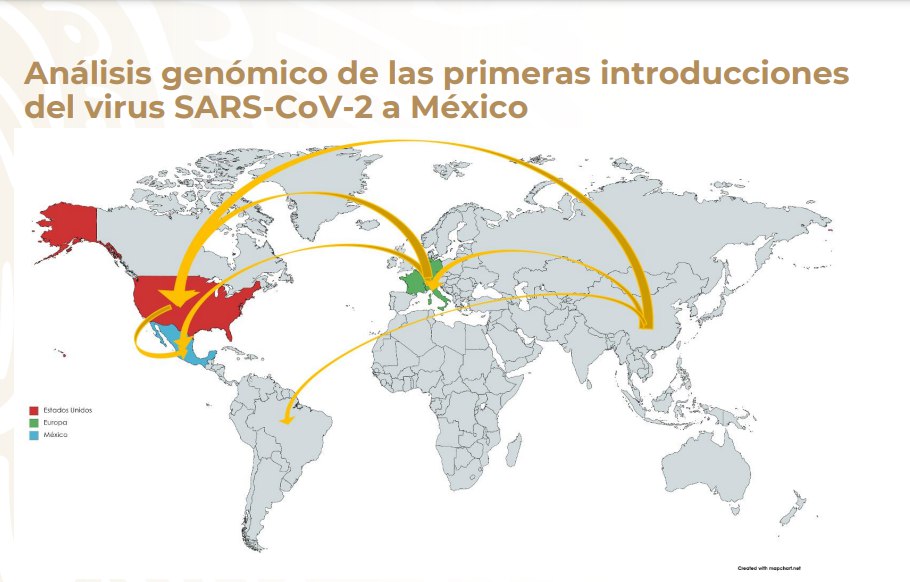
La secuencia genética de 17 genomas de SARS-CoV-2 confirmó que el virus que llegó a México era de un grupo distinto al que se esparció en China y otros países de Asia. El grupo del virus que llegó al país y que se confirmó el 27 de febrero provenía de Italia y había hecho escala en varios países de Europa, donde tuvo una mutación de la cepa original.
El virus también entró al país por Estados Unidos. En ambos casos, tanto el que venía de Italia como el de Estados Unidos, tocaron tierra en Ciudad de México y de ahí se dispersaron hacia el resto del país.
Éstas son las primeras conclusiones públicas del rastreo que han hecho los científicos mexicanos para conocer el comportamiento del virus que ha dejado, hasta este 22 de septiembre, más de 74 mil muertes en el país.
Este martes, en la conferencia diaria sobre covid-19, el director de la Unidad de Desarrollo Tecnológico en Investigación del Instituto de Investigación y Referencia Epidemiológica (Indre) Ernesto Ramírez González, presentó los resultados a los que han llegado investigadores de siete instituciones (Imegen, INER, INNSZ, UNAM, IMSS, Indre y Red Nacional de Laboratorios de Salud Pública) después de hacer asociaciones filogenéticas entre los virus presentes en México y en otras partes del mundo.
Para hacer estas asociaciones, el Indre reconstruyó la secuencia genética, el plano genético, de cientos de muestras de la enfermedad en el país y las comparó con secuencias genéticas de muestras de otros países.
“Nuestra primera introducción no venía de China como todo mundo creíamos, realmente la primera introducción vino de un viaje, de una introducción de Italia”, explicó Ramírez. “De China, la cepa primero hizo una escala en Europa, principalmente en países como Holanda, Italia y España, ahí se empezó a dispersar este virus y posteriormente ingresó a nuestro país”.
¿Para qué sirve la secuenciación?
Ramírez González explicó que, además de identificar el origen de una enfermedad, la secuenciación genética tiene distintas utilidades prácticas, como la identificación y clasificación de las diferentes mutaciones de virus que existen. Esta clasificación puede seguir dos grandes parámetros: el de los grupos o clados y el de linajes. Mientras sólo existen seis grupos (G, GH, GR, L, V y S), los linajes del virus son mucho más diversos y complejos, tanto que el número aumenta continuamente.
La secuenciación, identificación y clasificación de estas mutaciones permite la elaboración de vacunas, o el rastreo de inmunidad de alguna mutación de un virus a algún tratamiento. Por ejemplo, en el caso de la influenza H1N1, identificar estas mutaciones genéticas permitió observar qué mutación era resistente al oseltamivir, el medicamento que se usaba para tratar la enfermedad, comercializado comúnmente como tamiflú.
El proceso
Ramírez González explicó que durante los primeros 15 días después del primer caso de coronavirus en México, los investigadores empezaron a recolectar muestras de la enfermedad.
“Obtuvimos, de 33 muestras que seleccionamos, la secuencia de 17 genomas y con eso fue suficiente para darnos cuenta que estábamos teniendo dos vías de introducción a nuestro país, no sólo por Europa. Era por Europa y también por Estados Unidos”.
Incluso, estas primeras secuenciaciones, que es una manera de identificar qué mutaciones de las más de 18 mil en las que ha derivado el SARS-CoV-2, circulan en el país, le permitió a las autoridades sanitarias del país confirmar que la mayoría de los casos ingresaron primero a Ciudad de México a través del aeropuerto y de ahí se trasladaron a otros estados.
“Fue entonces que empezamos a hacer una cosa que se llama vigilancia genómica, ¿qué cepas?, ¿qué linajes?, ¿qué clados?, ¿qué está circulando en nuestro país o que se está presentando en nuestro país?” dijo al respecto.
No es el mismo virus que el de Asia
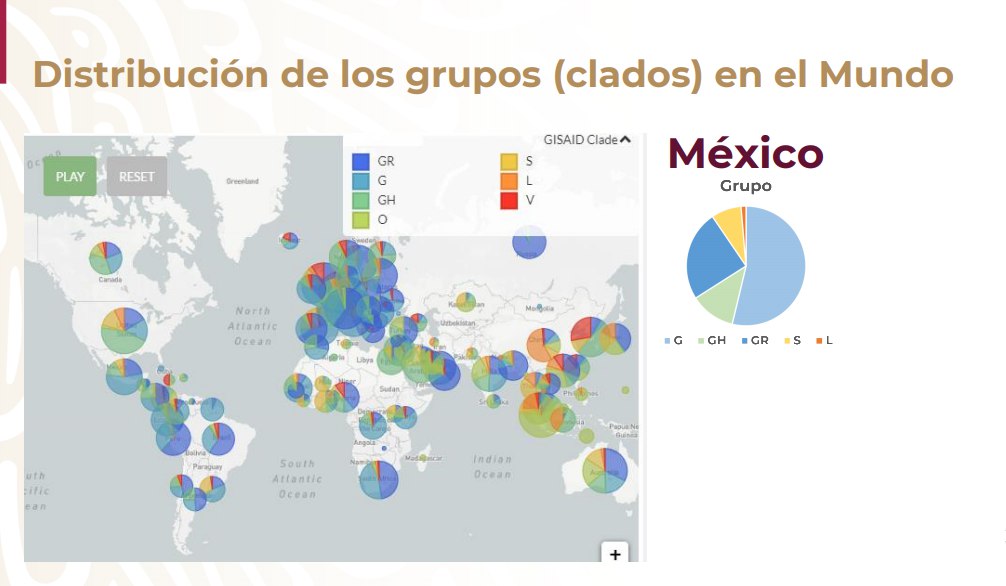
A partir de la secuenciación genética, el Indre logró identificar que en el país circulan cinco grupos diferentes de SARS-CoV-2, según su proporción, los más comunes son los de los grupos G, GH y GR, mutaciones que están en prácticamente todos los países de América y Europa (ahí predomina, sobre todo, el grupo GR).
En cambio, en Asia, se observa mayor presencia de los grupos S, L y V. Estas variaciones estuvieron presentes en México sólo al inicio de la pandemia.
“Tenemos la presencia de cinco grupos y 17 linajes de nuestro país, los grupos S y L que son más los grupos asiáticos sólo se presentaron prácticamente al inicio de la pandemia (…) en México predomina los grupos G, GH y GR que esto coincide con lo que se presenta en América y en Europa, y la presencia de estos linajes mayormente en América que son estos B.1 y todos los que están aquí”, explicó.
Esta posible relación fue adelantada hace unas semanas en una investigación especial de Pie de Página
Ramírez González explicó que, hasta ahora, no se ha logrado determinar que esta relación signifique una mayor severidad de la epidemia.
“Estamos trabajando fuertemente con los institutos. Tenemos 70 genomas ya ensamblados y estamos viendo si existiera alguna relación entre la severidad. A nivel internacional, hasta el momento, no se ha reportado que exista una mutación o algún marcador molecular que esté asociado con severidad; sin embargo, este resultado lo tendremos posteriormente”.
Relación con comorbilidades
Ramírez González destacó que los institutos mexicanos tienen las capacidades para poder llevar a cabo la obtención de estas secuencias.
“Es una aportación que estamos haciendo no sólo para nuestro país, estamos aportando información de calidad a nivel internacional, a nivel mundial, porque todas estas secuencias que quedan depositadas en fuentes públicas son consultadas por diferentes investigadores y ellos pueden hacer sus estudios y sus análisis”, explicó.
Por el momento, dijo, se está estudiando la relación de estos grupos con las comorbilidades. Además, están en marcha dos proyectos, uno con institutos de San Diego para estudiar el flujo e intercambio de grupos en la frontera norte, y otro con la Organización Panamericana de la Salud, para vigilar la frontera sur “porque no sabemos todavía si tenemos introducciones vía Sudamérica”.
“Queremos secuenciar mil genomas para tener un análisis más robusto y tener una mejor claridad de qué es lo que está ocurriendo en nuestro país”, dijo el científico.
También recomendamos:

Periodista en constante búsqueda de la mejor manera de contar cada historia y así dar un servicio a la ciudadanía. Analizo bases de datos y hago gráficas; narro vivencias que dan sentido a nuestra realidad.

Quería ser exploradora y conocer el mundo, pero conoció el periodismo y prefirió tratar de entender a las sociedades humanas. Dirigió seis años la Red de Periodistas de a Pie, y fundó Pie de Página, un medio digital que busca cambiar la narrativa del terror instalada en la prensa mexicana. Siempre tiene más dudas que respuestas.

Foránea siempre, lo suyo es lo audiovisual y el periodismo es la vía por donde conoce y cuestiona al mundo.
Ayúdanos a sostener un periodismo ético y responsable, que sirva para construir mejores sociedades. Patrocina una historia y forma parte de nuestra comunidad.
Dona














